



开放科学(资源服务)标识码(OSID)的开放科学计划以二维码为入口,提供丰富的线上扩展功能,包括作者对论文背景的语音介绍、该研究的附加说明、与读者的交互问答、拓展学术圈等。读者“扫一扫”此二维码即可获得上述增值服务。
异烟肼通过抑制细菌细胞壁的合成,能有效地阻止结核分枝杆菌的生长和繁殖,作为一线抗结核药品被广泛应用于结核病的治疗。异烟肼的代谢被认为与异烟肼介导的肝损伤相关。异烟肼通过氧化代谢途径产生的乙酰肼和肼为其主要的毒性代谢物,它们较母药更易造成肝损伤,其体内暴露量与肝损伤明显相关[1]。因此,对异烟肼及其代谢物乙酰肼和肼进行血药浓度监测,有助于实施临床个体化给药,减少药物不良反应的发生。
目前,检测肼类化合物的方法主要有气相色谱法[2]、高效液相色谱法(HPLC)[3]、气相色谱-质谱法[4]和高效液相色谱-质谱联用法(HPLC-MS/MS)[5⇓⇓-8]。临床应用中,常用于检测异烟肼血药浓度的方法主要是HPLC[9-10]。相较于其他方法,HPLC-MS/MS在靶向定量分析领域具有高敏感度、高选择性、宽线性范围、高准确性和重现性等显著优势。依托质谱技术,HPLC-MS/MS能够准确识别目标化合物并进行定量分析,适用于复杂样品的分析,并拓宽了仪器分析范围,为临床治疗药物监测提供了可靠的数据支持,被广泛应用于药物代谢动力学、生物样品分析等领域[11⇓⇓-14]。但已报道的方法存在前处理流程复杂、不能实现异烟肼及其代谢物同时检测等问题。因此,仍需要探索更简单灵敏的方法,可以同时测定人体血浆中的异烟肼及其代谢物乙酰肼和肼。本研究基于HPLC-MS/MS技术,以异烟肼-D4作为同位素标记内标,建立了快速、灵敏,可同时检测血浆中异烟肼及其代谢物乙酰肼、肼浓度的分析方法,并对校准曲线、线性范围、准确度、精密度、回收率、基质效应、稳定性进行了方法验证;同时,应用该方法对临床104例肺结核患者进行了异烟肼及其代谢物的治疗药物监测,以期为临床用药提供指导。
材料和方法
一、材料
1.仪器:UltraLC 110 液相串联API 4000 Q-trap型液-质联用系统(美国AB Sciex公司);电子分析天平(德国赛多利斯公司,BS110S);漩涡混合器(上海清甫仪器有限公司,SHA-C型);高速冷冻离心机(美国赛默飞世尔科技公司FRESCO21型离心机);超声波水浴清洗机(昆山市超声仪器有限公司)。
2.试剂:异烟肼(美国Sigma公司,批号:MKBW9046V);乙酰肼[梯希爱(上海)化成工业发展有限公司,批号:P8GWL-NT];肼(美国Sigma公司,批号:MKBV0553V);异烟肼-D4(南京昊绿生物科技有限公司,批号:YYJ0434314TR-HLM);色谱纯甲醇(美国赛默飞世尔科技公司,批号:204132);色谱纯乙腈(美国赛默飞世尔科技公司,批号:F21LA7202);对甲基苯甲醛(上海麦克林生化科技有限公司,批号:C10298201);甲酸(北京迪科马科技有限公司,批号:2142969);甲酸铵(美国Honeywell公司,批号:I2740);乙酸(上海吉至生化科技有限公司,批号:A331384C9)。
3.样品来源:患者来源于首都医科大学附属北京胸科医院,年龄范围18~60岁,确诊肺结核,并接受规范抗结核治疗。服用异烟肼的剂量为1次/d,每次300~500mg,于服药后2h采集静脉血4ml,4℃条件下,3000×g离心10min,取上层血浆放入-80℃冰箱内保存备用。共收集104例肺结核患者的血浆样本。本研究得到了首都医科大学附属北京胸科医院伦理委员会的批准(批准号:YJS-2022-16号),所有受试者在试验前均签署了知情同意书。
二、方法
1.色谱条件:采用InfinityLab Poroshell 120 HILIC-Z色谱柱(100mm×2.1mm,2.7μm);柱温为30℃;自动进样器温度为4℃;流动相A为含0.1%甲酸和5mmol甲酸铵的水溶液,流动相B为乙腈;梯度洗脱:0~2min,95% A;2~3min,95% A~5% A;3~5.5min,5% A;5.5~6.5min,5% A~95% A;6.5~8min,95% A;运行时间8min;进样量5μl;流速0.4ml/min。
2.质谱条件:质谱采用电喷雾离子源(ESI),以多反应监测(multiple reaction monitoring,MRM)模式扫描,正离子模式检测。电喷雾电压:5500V;离子源温度:500℃;入口电压、出口电压分别设置为10V、10V;气帘气(CUR):30psi;雾化气(GS1):50psi;加热气(GS2):50psi。待测物及内标异烟肼-D4衍生物的MRM参数见表1。
表1 高效液相色谱-质谱联用法的多反应监测参数设置
| 化合物 | 母离子(m/z) | 子离子(m/z) | 驻留时间(ms) | 碎裂电压(eV) | 去簇电压(eV) | 保留时间(min) |
|---|---|---|---|---|---|---|
| 异烟肼 | 240.0 | 118.0 | 100 | 23.0 | 65.0 | 3.65 |
| 乙酰肼 | 177.0 | 118.0 | 100 | 18.9 | 49.0 | 3.69 |
| 肼 | 237.3 | 119.0 | 100 | 28.8 | 55.0 | 4.24 |
| 异烟肼-D4 | 244.0 | 127.0 | 100 | 27.0 | 85.0 | 3.66 |
注 本参数设置表中的母离子和子离子信号均基于待测物与内标物和对甲基苯甲酸的衍生化产物的特征
3.溶液配制:(1)精密称取异烟肼、乙酰肼、肼的对照品适量,用甲醇分别配制成浓度均为1mg/ml的单一成分标准品储备液,-80℃避光保存。(2)精密量取异烟肼-D4溶液100μl于25ml量瓶中,用甲醇定容,即得质量浓度为100ng/ml的内标工作溶液。(3)精密量取对甲基苯甲醛2ml于100ml量瓶中,加入9.8ml乙酸,用甲醇定容,即得质量浓度为20μg/ml的对甲基苯甲醛溶液。
4.标准溶液配制:以甲醇稀释待测物储备液,配制系列浓度的线性混标工作溶液,混标工作溶液Line 1~Line 8中各待测物浓度分别为:(1)异烟肼:1、2、5、10、20、60、100、120μg/ml;(2)乙酰肼:0.5、1、2.5、5、10、30、50、60μg/ml;(3)肼:20、40、100、200、400、1200、2000、2400ng/ml。
取空白血浆95μl,加入5μl混标工作液Line 1~Line 8,各待测物在血浆中分别为:(1)异烟肼:50、100、250、500、1000、3000、5000、6000ng/ml;(2)乙酰肼:25、50、125、250、500、1500、2500、3000ng/ml;(3)肼:1、2、5、10、20、60、100、120ng/ml。
5.质控样本工作液配制:各待测物浓度分别为:(1)异烟肼:1、3、50、90μg/ml;(2)乙酰肼:0.5、1.5、25、45μg/ml;(3)肼:20、60、1000、1800ng/ml。
取空白血浆95μl,加入5μl混标工作液,各待测物在血浆中浓度分别为:(1)异烟肼:50、150、2500、4500ng/ml;(2)乙酰肼:25、75、1250、2250ng/ml;(3)肼:1、3、50、90ng/ml。
6.血浆样品处理:将待分析的血浆样品在室温下解冻,解冻完全后使用涡旋混合器将样品充分混匀。取100μl血浆加入350μl甲醇,50μl内标异烟肼-D4储备液(100ng/ml),涡旋混匀后,12000×g离心10min,取上清100μl加入300μl衍生化试剂对甲基苯甲醛溶液,充分混匀后室温超声反应40min,取200μl置于进样瓶中,上机进样分析。
三、统计学处理
采用SPSS 26.0软件,对数据进行描述性分析。计量资料呈正态分布,以“$\bar{x}±s$”描述。
结果
一、方法建立及验证
1.专属性考察:通过对比空白血浆基质、空白血浆基质+待测物混合对照品工作溶液(定量限浓度)+内标和临床患者血浆+内标的MRM色谱图,判断体内存在的其他内源性物质是否影响待测物的浓度测定,考察HPLC-MS/MS分析方法的专属性。结果显示,在本实验确定的检测条件下,样品中的内源性物质不影响待测物及内标的分离测定,且待测物与内标峰形良好。待测物与内标的MRM色谱图见图1。
图1
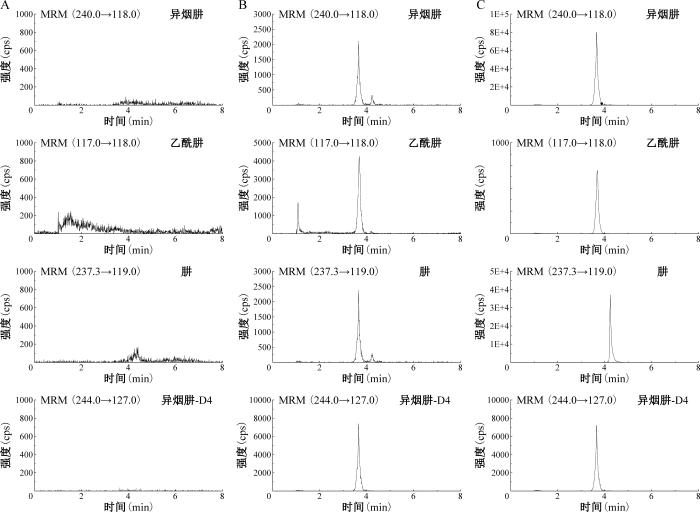
图1
高效液相色谱-质谱联用法多反应监测色谱图
注 图A:空白血浆;图B:空白血浆+待测物混合对照品工作溶液(定量限浓度)+内标;图C:临床患者血浆+内标;MRM:多反应监测
2.线性范围考察:配制每个浓度的标准曲线样品,进行处理后,进样分析,记录色谱图、待测物的峰面积(As)及内标的峰面积(Ai)。以浓度(C)为横坐标,待测物与内标峰面积的比值(As/Ai)为纵坐标作回归方程,权重系数为1/X2,结果如表2所示。异烟肼在线性范围50~6000ng/ml内线性良好,乙酰肼在线性范围25~3000ng/ml内线性良好,肼在线性范围1~120ng/ml内线性良好,决定系数(R2)均>0.99。
表2 高效液相色谱-质谱联用法测定异烟肼、乙酰肼和肼的线性范围、决定系数和定量限
| 化合物 | 回归方程 | 决定系数 (R2) | 线性范围 (ng/ml) |
|---|---|---|---|
| 异烟肼 | y=0.00548x-0.0004516 | 0.99974 | 50~6000 |
| 乙酰肼 | y=0.02353x-0.07664 | 0.99914 | 25~3000 |
| 肼 | y=0.31689x+0.23540 | 0.99949 | 1~120 |
3.残留:制备异烟肼、乙酰肼、肼定量上限(ULOQ)样品,质量浓度分别为4500、2250、90ng/ml。在每条标准曲线ULOQ样品后立即进行空白样品分析,考察高浓度样品残留对测定的影响从而计算残留效应。结果表明,空白样品中异烟肼、乙酰肼、肼的信号响应强度分别不超过定量下限(LLOQ)的0.12%、1.33%和2.06%,异烟肼-D4的信号响应强度不超过LLOQ的0.06%。
4.准确度与精密度:制备4个浓度的标准血浆样品溶液各6份,进行HPLC-MS/MS分析,考察分析方法的批内精密度和准确度。在不同时间连续制备并测定3个分析批次,考察方法的批间精密度和准确度。精密度用质控样品的批内和批间相对标准差(relative standard deviation,RSD)表示,准确度用相对误差(relative error,RE)表示。结果显示,异烟肼、乙酰肼、肼的批内和批间精密度RSD均≤11.76%,准确度RE绝对值均≤6.43%,符合生物样品检测的限度要求(精密度RSD<15%,准确度RE绝对值<15%)。具体见表3。
表3 高效液相色谱-质谱联用法测定化合物的准确度和精密度结果
| 化合物及 浓度 (ng/ml) | 日内(6份) | 日间(6份) | ||||
|---|---|---|---|---|---|---|
| 日内实测值 ($\bar{x}±s$,ng/ml) | 相对误差 (%) | 相对标准 差(%) | 日间实测值 ($\bar{x}±s$,ng/ml) | 相对误差 (%) | 相对标准 差(%) | |
| 异烟肼 | ||||||
| 50 | 50.97±1.28 | 1.93 | 2.49 | 50.35±4.40 | 0.70 | 8.74 |
| 150 | 143.03±2.46 | -4.65 | 1.91 | 144.84±8.11 | -3.44 | 5.60 |
| 2500 | 2339.24±38.19 | -6.43 | 1.44 | 2351.34±184.65 | -5.95 | 7.85 |
| 4500 | 4343.54±66.61 | -3.48 | 1.55 | 4276.56±273.83 | -4.97 | 6.40 |
| 乙酰肼 | ||||||
| 25 | 24.43±0.62 | -2.29 | 1.57 | 25.18±1.87 | 0.73 | 7.44 |
| 75 | 73.45±3.47 | -2.07 | 4.74 | 74.04±4.31 | -1.28 | 5.83 |
| 1250 | 1246.71±26.09 | -0.26 | 2.34 | 1203.44±81.71 | -3.72 | 6.79 |
| 2250 | 2272.32±27.16 | 0.99 | 1.34 | 2173.46±153.35 | -3.40 | 7.06 |
| 肼 | ||||||
| 1 | 0.99±0.08 | -1.00 | 7.05 | 1.02±0.09 | 1.43 | 11.76 |
| 3 | 3.08±0.12 | 2.53 | 3.90 | 3.03±0.24 | 0.96 | 7.94 |
| 50 | 51.67±2.76 | 3.33 | 5.81 | 48.96±2.91 | -2.08 | 5.95 |
| 90 | 93.48±1.94 | 3.86 | 2.32 | 88.87±5.23 | -1.25 | 5.89 |
5.回收率和基质效应:取空白血浆100μl,处理操作后得到空白基质,加入混合对照品工作溶液5μl,制备未经提取的低、中、高3个浓度的对照样品各6份,制备供试溶液,记录各待测物峰面积(At);取空白血浆95μl,加入混合对照品工作溶液5μl,配制成低、中、高浓度的标准血浆样品各6份,进行前处理,记录各待测物峰面积(AS);用起始比例流动相代替空白血浆,同上述方法制备相应浓度的溶液各6份,记录各待测物峰面积(AB)。
表4 高效液相色谱-质谱联用法测定化合物的提取回收率
| 化合物理论浓度 (ng/ml) | 提取回收率 ($\bar{x}±s$,%) | 相对标准差 (%) |
|---|---|---|
| 异烟肼 | ||
| 150 | 97.07±4.88 | 5.03 |
| 2500 | 100.59±3.58 | 3.56 |
| 4500 | 98.40±4.70 | 4.78 |
| 乙酰肼 | ||
| 75 | 94.66±3.13 | 3.31 |
| 1250 | 104.72±4.34 | 4.15 |
| 2250 | 100.77±5.38 | 5.33 |
| 肼 | ||
| 3 | 91.21±5.49 | 6.02 |
| 50 | 100.94±5.05 | 5.00 |
| 90 | 101.71±4.80 | 4.71 |
表5 高效液相色谱-质谱联用法测定化合物的基质效应结果
| 化合物理论浓度 (ng/ml) | 内标归一化基质 效应($\bar{x}±s$,%) | 相对标准差 (%) |
|---|---|---|
| 异烟肼 | ||
| 150 | 99.11±6.43 | 6.49 |
| 2500 | 105.95±6.01 | 5.68 |
| 4500 | 103.06±5.77 | 5.60 |
| 乙酰肼 | ||
| 75 | 101.09±5.41 | 5.35 |
| 1250 | 94.25±8.20 | 8.70 |
| 2250 | 103.80±4.87 | 4.69 |
| 肼 | ||
| 3 | 96.74±8.88 | 9.18 |
| 50 | 95.24±5.43 | 5.70 |
| 90 | 93.86±5.72 | 6.10 |
6.稳定性:取空白血浆配制低、中、高质控样品各3份,分别考察含药血浆样品室温放置24h、4℃冷藏24h、-80℃冻存7d、-80℃冻存14d、反复冻融3次的稳定性及血浆样品处理后在进样仓放置24h的稳定性。结果表明,待测物在各种条件下放置后,浓度无明显变化,说明在上述条件下稳定性良好(表6)。
表6 高效液相色谱-质谱联用法测定化合物浓度的稳定性
| 化合物浓度 (ng/ml) | 短期稳定性 (24h,25℃) | 短期稳定性 (24h,4℃) | 冻融稳定性 (3次冻融循环) | 长期稳定性 (14d,-80℃) | 进样器稳定性 (24h,10℃) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 相对误差 (%) | 变异系数 (%) | 相对误差 (%) | 变异系数 (%) | 相对误差 (%) | 变异系数 (%) | 相对误差 (%) | 变异系数 (%) | 相对误差 (%) | 变异系数 (%) | |
| 异烟肼 | ||||||||||
| 150 | -0.38 | 1.49 | -3.30 | 2.19 | -4.69 | 6.37 | -5.51 | 4.03 | -7.67 | 1.62 |
| 2500 | -1.25 | 1.09 | -1.35 | 1.72 | -4.53 | 0.40 | -4.01 | 1.06 | -6.67 | 2.34 |
| 4500 | 3.48 | 1.02 | 5.46 | 0.97 | 1.18 | 0.68 | 0.53 | 0.55 | -5.38 | 9.77 |
| 乙酰肼 | ||||||||||
| 75 | 6.61 | 5.29 | 4.59 | 5.39 | -0.53 | 1.64 | 0.88 | 0.22 | -7.86 | 0.85 |
| 1250 | 4.55 | 4.56 | 7.88 | 1.68 | 7.74 | 1.09 | 6.04 | 3.72 | -7.03 | 3.04 |
| 2250 | 5.41 | 5.88 | 0.12 | 0.68 | 1.79 | 6.16 | -1.65 | 1.02 | -3.56 | 2.64 |
| 肼 | ||||||||||
| 3 | -6.59 | 4.31 | -5.69 | 2.44 | -1.31 | 5.51 | 0.96 | 5.87 | -5.21 | 3.27 |
| 50 | -4.04 | 4.18 | -6.58 | 2.08 | 6.80 | 2.13 | 6.87 | 3.26 | -7.79 | 3.32 |
| 90 | -5.31 | 5.13 | -7.18 | 6.34 | -4.69 | 6.37 | 3.51 | 0.92 | -7.36 | 6.27 |
二、临床应用
用上述方法检测104例肺结核患者血浆中异烟肼及其代谢物。根据随行标准曲线计算样本中待测物的浓度。异烟肼的浓度范围为359.41~5866.71ng/ml,乙酰肼的浓度范围为667.11~2997.79ng/ml,肼的浓度范围为4.38~112.66ng/ml(表7)。
表7 高效液相色谱-质谱联用法检测临床血浆样本中异烟肼、乙酰肼、肼的浓度
| 化合物 | 浓度范围 (ng/ml) | 浓度 ($\bar{x}±s$,ng/ml) | 相对标准差 (%) |
|---|---|---|---|
| 异烟肼 | 359.41~5866.71 | 2402.33±1248.57 | 51.97 |
| 乙酰肼 | 667.11~2997.79 | 1902.51±596.82 | 31.37 |
| 肼 | 4.38~112.66 | 26.50±17.13 | 64.62 |
讨论
本研究建立的HPLC-MS/MS法具有以下优势:(1)本方法不仅可以检测异烟肼,还能同时分析与药物性肝损伤有关的代谢物乙酰肼和肼,为异烟肼的治疗药物监测提供了更全面的支持。(2)本方法采用同位素内标具有出色的分辨率和高度准确的检测能力,能够有效地分离和鉴定异烟肼及其代谢物,从而确保结果的准确性和可靠性。(3)本方法能够在更宽的浓度范围内检测异烟肼及其代谢物,提高了方法的适用性和可靠性。
由于乙酰肼和肼的弱保留性,通常很难通过HPLC-MS/MS直接检测,故采用衍生化法对乙酰肼和肼进行前处理后进行分析,以提高检测性能。文献中报道的异烟肼及其代谢物的血药浓度测定方法将异烟肼与乙酰肼和肼分开检测,采用不同的前处理方法和分析方法,这大大增加了前处理及分析的流程和时间[8]。本研究将异烟肼、乙酰肼和肼同时进行衍生化,成功实现了异烟肼及其代谢物乙酰肼和肼在同一前处理条件下的同时测定。
本研究选择对甲基苯甲醛为衍生化试剂,对待测物进行衍生化,并对衍生化条件进行了摸索和优化;选择室温超声作为反应条件,考察了不同反应时间(10、20、30、40、50min)对信号响应的影响,选择最佳反应时间为40min。针对待测物经对甲基苯甲醛衍生化后,在HPLC-MS/MS分析时存在衍生反应还未到反应终点问题,影响了定量结果的准确性和稳定性的问题,本研究采取加大衍生化试剂对甲基苯甲醛的用量,并于衍生化反应体系中加入适量乙酸,使反应进行完全。在反应过程中引入超声波以提高分子碰撞的概率,从而提高反应产率。与加热衍生化方法相比,超声水浴可以获得微米和纳米级的聚集体,缩短反应时间,提高反应效率[21]。通过使用超声波,本研究的样品预处理流程简单,无需额外的加热和纯化程序。
异烟肼是目前广泛应用于结核病治疗的抗结核药物之一。然而,其代谢物存在潜在的肝毒性,这一点在临床应用中需要引起高度重视。本研究检测了异烟肼给药后2h临床血浆样本中异烟肼及其代谢物乙酰肼和肼的浓度水平,结果显示,患者体内的异烟肼和代谢物乙酰肼和肼的浓度存在较大的个体间差异。Song等[7]检测了异烟肼末次给药24h后患者体内异烟肼及肼的浓度,结果表明,长期服用异烟肼后,患者体内残留有乙酰肼和肼。而乙酰肼和肼的残留量在不同患者中差异较大。因此,需要对临床服用异烟肼的患者进行血药浓度监测,对于血药浓度明显增高的患者,需要密切关注其药物不良反应情况;而对于血药浓度偏低的患者,建议医生调整给药剂量。临床应结合患者的个体特征和治疗目标,从疗效和肝毒性两方面考虑,制订最佳的个体化给药方案,适当调整异烟肼剂量,以达到最佳的治疗效果。
综上所述,本研究建立了一种快速灵敏的HPLC-MS/MS方法,可用于测定人体内异烟肼及其代谢物乙酰肼、肼的血药浓度,通过对方法专属性、标准曲线、精密度、准确度、基质效应、回收率和稳定性等的验证,本研究建立的方法准确度高、专属性强,已应用于异烟肼及其代谢产物的临床监测。该方法有助于临床优化异烟肼的个体化给药,从而减少异烟肼引起的肝损伤的发生。
利益冲突 所有作者均声明不存在利益冲突
作者贡献 葛菲:酝酿和设计实验、实施研究、起草文章、统计分析;朱慧:采集数据、分析/解释数据、对文章的知识性内容作批评性审阅、指导;程凯:分析/解释数据、对文章的知识性内容作批评性审阅、支持性贡献;陆宇:分析/解释数据、对文章的知识性内容作批评性审阅、行政/技术/材料支持、指导;徐建:酝酿和设计实验、分析/解释数据、对文章的知识性内容作批评性审阅、获取研究经费、行政/技术/材料支持、指导
参考文献
Isoniazid metabolism and hepatotoxicity

Isoniazid (INH) is highly effective for the management of tuberculosis. However, it can cause liver injury and even liver failure. INH metabolism has been thought to be associated with INH-induced liver injury. This review summarized the metabolic pathways of INH and discussed their associations with INH-induced liver injury.
Control and analysis of hydrazine, hydrazides and hydrazones--genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products
This is the latest of a series of reviews focused on the analysis of genotoxic impurities. This review summarises the analytical approaches reported in the literature relating to hydrazine, hydrazines, hydrazides and hydrazones. It is intended to provide guidance for analysts needing to develop procedures to control such impurities, particularly where this is due to concerns relating to their potential genotoxicity. Of particular note is the wide variety of techniques employed, both chromatographic and spectroscopic, with most involving derivatisation. Such a wide variety of options allow the analyst a real choice in terms of selecting the most appropriate technique specific to their requirements. Several generic methodologies, covering the three main analytical approaches; i.e. HPLC (high performance liquid chromatography), GC (gas chromatography) and IC (ion chromatography), are also described.Copyright © 2010 Elsevier B.V. All rights reserved.
Direct liquid chromatographic determination of hydrazines: a review
Today the determination of hydrazines is an important application in analytical chemistry. This review shows the current state-of-the-art analyses and discusses the merits of the direct chromatographic methods for the determination of hydrazines such as ion-, ion-exclusion, ion-pair and hydrophilic interaction chromatography. The methodological aspects of the separation and detection of hydrazines are considered for these methods. Examples of hydrazine determination in real samples are presented.Copyright © 2012 Elsevier B.V. All rights reserved.
A generic approach for the determination of trace hydrazine in drug substances using in situ derivati-zation-headspace GC-MS
基于超高效液相色谱—质谱技术血浆神经酰胺含量分析
Quantification of Hydrazine in Human Urine by HPLC-MS-MS
Currently used on F-16 fighter jets and some space shuttles, hydrazine could be released at toxic levels to humans as a result of an accidental leakage or spill. Lower-level exposures occur in industrial workers or as a result of the use of some pharmaceuticals. A method was developed for the quantitation of hydrazine in human urine and can be extended by dilution with water to cover at least six orders of magnitude, allowing measurement at all clinically significant levels of potential exposure. Urine samples were processed by isotope dilution, filtered, derivatized and then quantified by HPLC-MS-MS. The analytical response ratio was linearly proportional to the urine concentration of hydrazine from 0.0493 to 12.3 ng/mL, with an average correlation coefficientRof 0.9985. Inter-run accuracy for 21 runs, expressed as percent relative error (% RE), was ≤14%, and the corresponding precision, expressed as percent relative standard deviation (% RSD), was ≤15%. Because this method can provide a quantitative measurement of clinical samples over six orders of magnitude, it can be used to monitor trace amounts of hydrazine exposure as well as industrial and environmental exposure levels. Published by Oxford University Press 2016. This work is written by (a) US Government employee(s) and is in the public domain in the US.
Simultaneous quantitation of hydrazine and acetylhydrazine in human plasma by high performance liquid chromatography-tandem mass spectrometry after derivatization with p-tolualdehyde
A high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method was developed for simultaneous quantitative analysis of hydrazine and acetylhydrazine in human plasma based on the strategy of p-tolualdehyde derivatization. The derivatization reactions were easily realized by ultrasonic manipulation for 40min. Good separation of the derivatization products was achieved using a C column by gradient elution. The optimized mass transition ion-pairs (m/z) monitored for the two hydrazine derivatives were m/z 237.1≫>119.9 and m/z 176.9≫>117.8, respectively. The limit of detection (LOD) and limit of quantification (LOQ) for hydrazine were 0.002 and 0.005ngmL separately. And they were 0.03 and 0.05ngmL for acetylhydrazine, respectively. The linear range was 0.005-50ngmL for hydrazine and 0.05-500ngmL for acetylhydrazine with R greater than 0.999. The recovery range was determined to be 95.38-108.12% with the relative standard deviation (RSD) in the range of 1.24-14.89%. The method was successfully applied to detect 30 clinical plasma samples of pulmonary tuberculosis patients treated with isoniazid. The concentrations were from 0.04-1.99ngmL for hydrazine and 0.06-142.43ngmL for acetylhydrazine. The results indicated that our developed method had the potential for the detection of hydrazine toxicology in complex biological samples. Furthermore, the method has an important significance to clinical treatment with drugs.Copyright © 2017. Published by Elsevier B.V.
Quantitative Analysis of Isoniazid and Its Four Primary Metabolites in Plasma of Tuberculosis Patients Using LC-MS/MS
Isoniazid and its metabolites are potentially associated with hepatotoxicity and treatment outcomes in patients who receive antituberculosis (TB) therapy. To further understand the pharmacokinetic profiles of these molecules, a method based on LC-MS/MS was developed to determine the concentration of these compounds in human plasma. Isoniazid, acetylisoniazid, and isonicotinic acid were directly analyzed, whereas hydrazine and acetylhydrazine were determined after derivatization using p-tolualdehyde. Chromatographic separation was conducted on reversed-phase C18 columns with gradient elution, and detection was carried out in multiple reaction monitoring mode. The calibration curves were linear with correlation coefficients (r) greater than 0.9947 for all analytes. The intra- and inter-day precision was less than 13.43%, and the accuracy ranged between 91.63 and 114.00%. The recovery and matrix effect of the analytes were also consistent (coefficient of variation was less than 9.36%). The developed method successfully quantified isoniazid and its metabolites in TB patients. The method has broad applications in clinical research, including isoniazid one-point-based therapeutic drug monitoring, genotype–phenotype association studies of isoniazid metabolic profile and isoniazid-induced hepatotoxicity, and the initial dose prediction of isoniazid using population pharmacokinetic modeling.
异烟肼血药浓度的影响因素分析
Development of a HPLC Method for Analysis of a Combination of Clofazimine, Isoniazid, Pyrazinamide, and Rifampicin Incorporated into a Dermal Self-Double-Emulsifying Drug Delivery System
We describe the development and validation of a new high performance liquid chromatography (HPLC) method for analysis of a combination of the first-line anti-tubercular drugs isoniazid, pyrazinamide, and rifampicin together with clofazimine. This is a unique challenge since clofazimine and rifampicin are relatively highly lipophilic drugs, whereas isoniazid and pyrazinamide are considerably more hydrophilic. Thus, clear separation of peaks and quantification of four individual drugs can present difficulties during the development of an analytical method. Detection was established at two wavelengths—254 nm for isoniazid and pyrazinamide and 320 nm for clofazimine and rifampicin. Gradient elution was employed using 0.1% aqueous formic acid (A) and acetonitrile (B); clear separation of the four drugs was achieved within 10 min. A linear relationship was indicated by a correlation coefficient (r2) of 0.9999 for each anti-tubercular drug, respectively. The limit of detection (LOD) for the individual drugs was 0.70 µg/mL (isoniazid), 0.30 µg/mL (pyrazinamide), 0.20 µg/mL (rifampicin) and 0.20 µg/mL (clofazimine). Precision experiments rendered a mean recovery percentage of 101.25% (isoniazid), 98.70% (pyrazinamide), 99.68% (rifampicin) and 97.14% (clofazimine). This HPLC method was validated and is reliable, repeatable, and accurate for the purpose of conducting simultaneous HPLC analyses of the four anti-tubercular drugs.
Cutting-edge LC-MS/MS applications in clinical mass spectrometry: Focusing on analysis of drugs and metabolites
In recent years, liquid chromatography with tandem mass spectrometry (LC–MS/MS) has become a fundamental technology in clinical practice. In Japan, the LC–MS/MS system is used in many large hospitals. It has become popular among pharmacists and laboratory technicians. LC–MS/MS has some advantages in terms of accuracy, speed, and comprehensiveness compared to conventional automated chemical testing equipment. However, LC–MS/MS is by no means a universal method, and it is necessary to understand its characteristics before using it. In the field of therapeutic drug monitoring (TDM), there is an issue with linearity in comprehensive measurement; however, ion‐abundance adjustment methods, such as in‐source collision‐induced dissociation, have been proposed as a solution to this problem. The development of a biomarker analysis includes search, identification, and quantification, and it is necessary to select an appropriate mass spectrometric method for each step. In this paper, we review cutting‐edge technologies that can expand the performance of LC–MS/MS in the clinical field and consider current issues and future prospects.
Advances in antifungal drug measurement by liquid chromatography-mass spectrometry
Fungal infections, especially invasive types, have become a serious healthcare problem as the immunocompromised population increases. There are five main classes of antifungal drugs: polyenes, flucytosine, allylamines, azoles, and echinocandins. Therapeutic drug monitoring (TDM) is justified for flucytosine and triazoles due to their large inter- and intra-individual pharmacokinetic variability and their high tendency for drug-drug interactions. Available methods for measuring these drugs include bioassay, liquid chromatography and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). The LC-MS/MS approach is preferred due to its superior analytic sensitivity and specificity. In this review, we highlight TDM methods by LC-MS/MS for these antifungal drugs searchable in PubMed by December 1, 2018. LC-MS/MS methods that were developed for other purposes such as pharmacokinetics or toxicokinetics were also included. We have critically analyzed these methods with an emphasis on sensitivity, specificity, simplicity, throughput and robustness.Copyright © 2019 Elsevier B.V. All rights reserved.
Liquid Chromatography-Tandem Mass Spectrometry: An Emerging Technology in the Toxicology Laboratory
In the last decade, liquid chromatography-tandem mass spectrometry (LC-MS/MS) has seen enormous growth in routine toxicology laboratories. LC-MS/MS offers significant advantages over other traditional testing, such as immunoassay and gas chromatography-mass spectrometry methodologies. Major strengths of LC-MS/MS include improvement in specificity, flexibility, and sample throughput when compared with other technologies. Here, the basic principles of LC-MS/MS technology are reviewed, followed by advantages and disadvantages of this technology compared with other traditional techniques. In addition, toxicology applications of LC-MS/MS for simultaneous detection of large panels of analytes are presented.Copyright © 2016 Elsevier Inc. All rights reserved.
Therapeutic drug monitoring and LC-MS/MS
Association of Cytochrome P450 2E1 and N-Acetyltransferase 2 Genotypes with Serum Isoniazid Level and Anti-Tuberculosis Drug-Induced Hepatotoxicity: A Cross-Sectional Study
Mechanisms of isoniazid-induced idiosyncratic liver injury: emerging role of mitochondrial stress
Idiosyncratic drug‐induced liver injury (DILI) is a significant adverse effect of antitubercular therapy with isoniazid (INH). Although the drug has been used for many decades, the underlying mode of action (both patient‐specific and drug‐specific mechanisms) leading to DILI are poorly understood. Among the patient‐specific determinants of susceptibility to INH‐associated DILI, the importance of HLA genetic variants has been increasingly recognized, whereas the role of polymorphisms of drug‐metabolizing enzymes (NAT2 and CYP2E1) has become less important and remains controversial. However, these polymorphisms are merely correlative, and other molecular determinants of susceptibility have remained largely unknown. Regarding the drug‐specific mechanisms underlying INH‐induced liver injury, novel concepts have been emerging. Among these are covalent protein adduct formation via novel reactive intermediates, leading to hapten formation and a potential immune response, and interference with endogenous metabolism. Furthermore, INH and/or INH metabolites (e.g. hydrazine) can cause mitochondrial injury, which can lead to mitochondrial oxidant stress and impairment of energy homeostasis. Recent studies have revealed that underlying impairment of complex I function can trigger massive hepatocellular injury induced by otherwise nontoxic concentrations of INH superimposed on these mitochondrial deficiencies. This review discusses these emerging new paradigms of INH‐induced DILI and highlights recent insights into the mechanisms, as well as points to the existing large gaps in our understanding of the pathogenesis.
The Isoniazid Metabo-lites Hydrazine and Pyridoxal Isonicotinoyl Hydrazone Modulate Heme Biosynthesis
Simultaneous determination of first-line anti-tuberculosis drugs and their major metabolic ratios by liquid chromatography/tandem mass spectrometry
Monitoring of anti‐tuberculosis drug concentrations and dose adjustment can be helpful in cases that show poor response to treatment. Here, we describe a method that can rapidly and simultaneously measure the blood concentrations of four anti‐tuberculosis drugs (isoniazid, rifampicin, pyrazinamide, and ethambutol) and two major metabolic ratios (acetylisoniazid/isoniazid and 25‐desacetylrifampicin/rifampicin) using high‐performance liquid chromatography/tandem mass spectrometry (HPLC/MS/MS). A C18 reversed‐phase column and gradients of methanol in 0.3% formic acid and water were used for HPLC separation. The drug concentrations were determined by multiple reaction monitoring in positive ion mode and the assay performance was evaluated. We determined peak concentration ranges for each drug and acetylisoniazid/isoniazid and 25‐desacetylrifampicin/rifampicin ratios by analyzing 2‐h post‐dose samples in patients treated with standard dosing as a first‐line treatment. The preparation of 20 samples including two steps of deproteinization with 50% and 100% methanol was performed within 20 min and chromatographic separation was achieved within 4 min/sample. Interassay calibration variability data obtained over concentrations of 0–8 µg/mL for isoniazid and ethambutol and 0–80 µg/mL for rifampicin and pyrazinamide showed a linear and reproducible curve. Within‐run and between‐run imprecision (CVs) were 1.9–5.5% and 3.5–10.5% and the lower limits of detection and quantification were 0.01–0.5 µg/mL and 0.05–1.0 µg/mL, respectively. The isoniazid concentration was found to be inversely correlated to the acetylisoniazid/isoniazid ratio (R = −0.739, P < 0.001). The devised method allows for the simple, rapid, sensitive and reproducible quantification of isoniazid, rifampicin, pyrazinamide, ethambutol and their two metabolic ratios and should be helpful for therapeutic drug monitoring in tuberculosis patients. Copyright © 2007 John Wiley & Sons, Ltd.
A battery of tandem mass spectrometry assays with stable isotope-dilution for the quantification of 15 anti-tuberculosis drugs and two metabolites in patients with susceptible-, multidrug-resistant- and extensively drug-resistant tuberculosis
Rapid and sensitive method for simultaneous determination of first-line anti-tuberculosis drugs in human plasma by HPLC-MS/MS: Application to therapeutic drug monitoring
Ultrasound assisted-deep eutectic solvent based on emulsification liquid phase microextraction combined with microsample injection flame atomic absorption spectrometry for valence speciation of chromium (Ⅲ/Ⅵ) in environmental samples
A new type of deep eutectic solvents (DESs) have been prepared and used as extraction solvents for ultrasound assisted-deep eutectic solvent based emulsification liquid phase microextraction method (UA-DES-ELPME) for the determination and speciation of total chromium, chromium(III) and chromium(VI). The chromium concentration in DES rich phase (extraction phase) was determined by using microsample injection flame atomic absorption spectrometer (FAAS). The detection limit (LOD), the quantification limit (LOQ), preconcentration factor and relative standard deviation were found as 5.5µgL(-1), 18.2µgL(-1), 20 and 6%, respectively. The accuracy of the developed method was evaluated by the analysis of water the certified reference materials (TMDA-53.3 Fortified environmental water and TMDA-54.4 Fortified Lake Water) and addition-recovery tests for water samples. Copyright © 2016 Elsevier B.V. All rights reserved.
Selecting a Structural Analog as an Internal Standard for the Quantification of 6-Methylmercaptopurine by LC-MS/MS
When choosing an analog internal standard (IS) in a quantitative LC-MS/MS assay, careful selection and thorough verification are important for developing an accurate quantitative assay. The IS is a critical component in quantitative mass spectrometry because it is used to normalize results by compensating for variations in sample preparation and instrument performance. Here we present the results of our investigation in the selection process for a structural analog IS (SA-IS) to be used in the quantification of 6-methylmercaptopurine (6-MMP) in cytolysed red blood cell (RBC).A cocktail solution of 9 SA-ISs including the isotopically labeled structural isomer and the 6-MMP stable isotope-labeled IS (SIL-IS) was spiked into cytolysed RBC controls and patient samples. Linearity, accuracy, sensitivity, precision, run stability, method comparison, and reinjection reproducibility experiments were performed. Ion suppression was also assessed by T-infusing the cocktail solution.All analogs were linear from 100 to 1200 ng/mL 6-MMP with acceptable precision and sensitivity by use of a spiked blank lysate. Method comparison plots of 6-MMP concentrations in patient samples had excellent agreement for 2 of the SA-ISs (i.e., the isotopically labeled structural isomer and an SA-IS with an added methyl group) when compared to the SIL-IS. Halogen-substituted analogs (i.e., Cl and Br) also met the criteria as an acceptable IS. However, 2 of the selected SA-ISs having substituted amine moieties showed unacceptable performance, with ≥15% bias when compared to the SIL-IS.There are many parameters to consider when determining if an analog will be a good IS choice, and the approaches highlighted in this article can be applied to the selection of SA-IS in the development of other LC-MS/MS assays.© 2018 American Association for Clinical Chemistry.
Diagnostic Accuracy of Therapeutic Drug Monitoring During Tuberculosis Treatment
Patients with tuberculosis (TB) coinfected with HIV are more likely to have low blood concentrations of the first‐line anti‐TB drugs (associated with poor outcomes). Therapeutic drug monitoring (TDM) is recommended for certain patient populations with TB at increased risk for a poor outcome. Our objective was to estimate the diagnostic accuracy of a 2‐hour TDM serum sample for the first‐line anti‐TB drugs among patients with HIV/TB and evaluate the information gained by an additional 6‐hour sample. We created a virtual (n = 1000) HIV/TB patient population and performed pharmacokinetic simulations using published population models for isoniazid, rifampin, pyrazinamide, and ethambutol. We performed receiver operating characteristic analysis to compare the diagnostic performance of a single 2‐hour serum sample with samples obtained at 2 and 6 hours after dosing. The sensitivity of a single 2‐hour serum concentration to identify patients with HIV/TB with adequate serum exposures was lowest for rifampin (54.9%; 95%CI, 50.79%‐59.41%) and highest for ethambutol (70.8%; 95%CI, 66.06%‐72.61%) for maximum concentration (Cmax) targets. Diagnostic accuracy of a single 2‐hour serum sample for the area under the concentration‐time curve (AUC) from time 0 to 24 hours target was highest for isoniazid (93%; 95%CI, 90.9%‐94.1%) and lowest for pyrazinamide (66.3%; 95%CI, 62.6%‐70.0%). In summary, the diagnostic performance of TDM for Cmax and AUC from time 0 to 24 hours targets demonstrated variability across the first‐line anti‐TB drugs. The addition of a 6‐hour serum sample led to the highest statistically significant improvement (P <.001) and highest increase in diagnostic accuracy (area under the receiver operating characteristic curve) for rifampin for Cmax and AUC. The other first‐line drugs had modest/negligible increases in diagnostic accuracy.
Optimizing treatment outcome of first-line anti-tuberculosis drugs: the role of therapeutic drug monitoring
Tuberculosis (TB) remains one of the world's deadliest communicable diseases. Although cure rates of the standard four-drug (rifampicin, isoniazid, pyrazinamide, ethambutol) treatment schedule can be as high as 95-98 % under clinical trial conditions, success rates may be much lower in less well resourced countries. Unsuccessful treatment with these first-line anti-TB drugs may lead to the development of multidrug resistant and extensively drug resistant TB. The intrinsic interindividual variability in the pharmacokinetics (PK) of the first-line anti-TB drugs is further exacerbated by co-morbidities such as HIV infection and diabetes.Therapeutic drug monitoring has been proposed in an attempt to optimize treatment outcome and reduce the development of drug resistance. Several studies have shown that maximum plasma concentrations (C max), especially of rifampicin and isoniazid, are well below the proposed target C max concentrations in a substantial fraction of patients being treated with the standard four-drug treatment schedule, even though treatment's success rate in these studies was typically at least 85 %.The proposed target C max concentrations are based on the concentrations of these agents achieved in healthy volunteers and patients receiving the standard doses. Estimation of C max based on one or two sampling times may not have the necessary accuracy since absorption rate, especially for rifampicin, may be highly variable. In addition, minimum inhibitory concentration (MIC) variability should be taken into account to set clinically meaningful susceptibility breakpoints. Clearly, there is a need to better define the key target PK and pharmacodynamic (PD) parameters for therapeutic drug monitoring (TDM) of the first-line anti-TB drugs to be efficacious, C max (or area under the curve (AUC)) and C max/MIC (or AUC/MIC).Although TDM of first-line anti-TB drugs has been successfully used in a limited number of specialized centers to improve treatment outcome in slow responders, a better characterization of the target PK and/or PK/PD parameters is in our opinion necessary to make it cost-effective.

 京公网安备11010202007215号
京公网安备11010202007215号
{kind=link}
{kind=link}