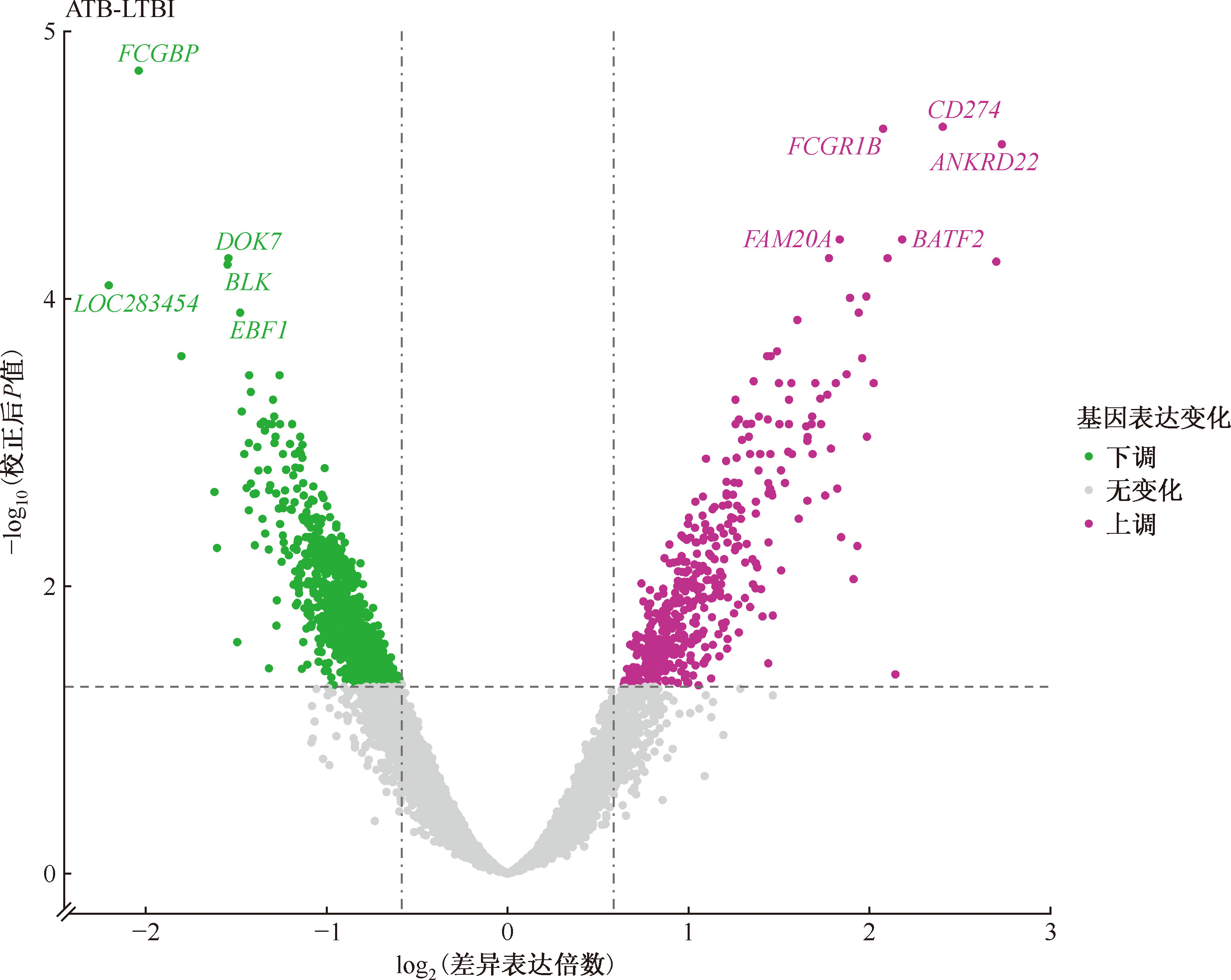
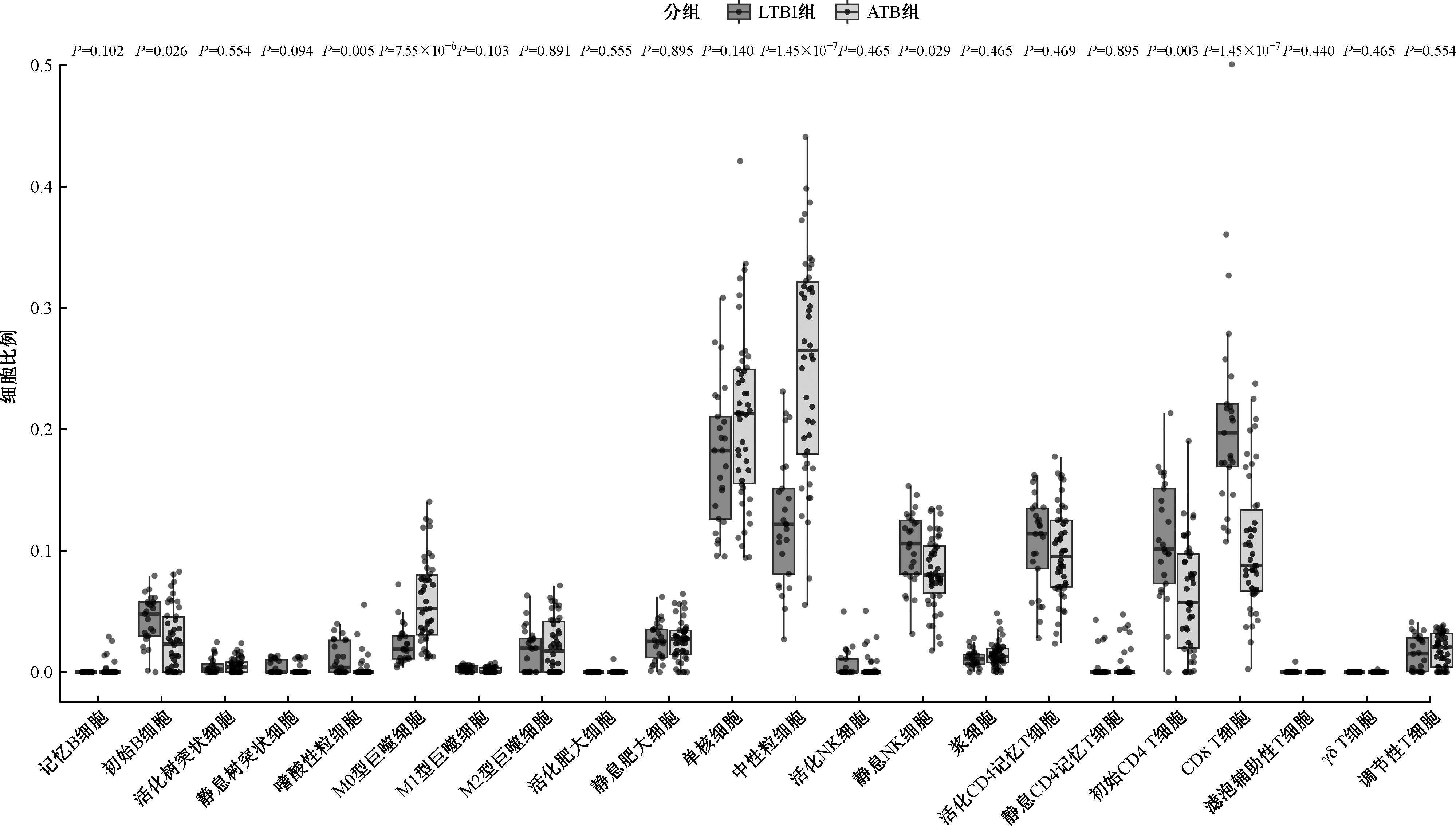
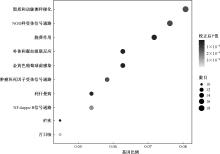
Chinese Journal of Antituberculosis ›› 2026, Vol. 48 ›› Issue (1): 139-147.doi: 10.19982/j.issn.1000-6621.20250352
• Original Articles • Previous Articles Next Articles
Fan Weixiao, Zhou Ke, Liu Jiayun( )
)
Received:2025-08-29
Online:2026-01-10
Published:2025-12-31
Contact:
Liu Jiayun, Email: Supported by:CLC Number:
Fan Weixiao, Zhou Ke, Liu Jiayun. Identification of efferocytosis-related core genes in active tuberculosis patients based on GEO database[J]. Chinese Journal of Antituberculosis, 2026, 48(1): 139-147. doi: 10.19982/j.issn.1000-6621.20250352
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.zgflzz.cn/EN/10.19982/j.issn.1000-6621.20250352
| [1] | 屈燕, 李涛, 马文斌, 等. 世界卫生组织《2025年全球结核病报告》解读. 结核与肺部疾病杂志, 2025, 6(6):613-623. doi:10.19983/j.issn.2096-8493.20250178. |
| [2] | Chen Z, Wang T, Du J, et al. Decoding the WHO Global Tuberculosis Report 2024: A Critical Analysis of Global and Chinese Key Data. Zoonoses, 2025, 5(1): 1-16. doi:10.15212/ZOONOSES-2024-0061. |
| [3] | 胡鑫洋, 高静韬. 世界卫生组织《2024年全球结核病报告》解读. 结核与肺部疾病杂志, 2024, 5(6): 500-504. doi:10.19983/j.issn.2096-8493.2024164. |
| [4] | 高磊, 张慧, 胡茂桂, 等. 基于多中心调查数据和空间统计模型的全国结核分枝杆菌潜伏感染率估算. 中国防痨杂志, 2022, 44(1): 54-59. doi:10.19982/j.issn.1000-6621.20210661. |
| [5] |
Hartman-Adams H, Clark K, Juckett G. Update on latent tuberculosis infection. Am Fam Physician, 2014, 89(11): 889-896.
pmid: 25077395 |
| [6] |
Mazurek GH, Jereb J, Vernon A, et al. Updated guidelines for using Interferon Gamma Release Assays to detect Mycobacterium tuberculosis infection-United States, 2010. MMWR Recomm Rep, 2010, 59(RR-5): 1-25.
pmid: 20577159 |
| [7] |
Chee CB, Gan SH, Khinmar KW, et al. Comparison of sensitivities of two commercial gamma interferon release assays for pulmonary tuberculosis. J Clin Microbiol, 2008, 46(6): 1935-1940. doi:10.1128/JCM.02403-07.
pmid: 18400912 |
| [8] | Nisa A, Kipper FC, Panigrahy D, et al. Different modalities of host cell death and their impact on Mycobacterium tuberculosis infection. Am J Physiol Cell Physiol, 2022, 323(5): C1444-C1474. doi:10.1152/ajpcell.00246.2022. |
| [9] | Zhuang L, Yang L, Li L, et al. Mycobacterium tuberculosis: immune response, biomarkers, and therapeutic intervention. MedComm (2020), 2024, 5(1): e419. doi:10.1002/mco2.419. |
| [10] | Warsinske H, Vashisht R, Khatri P. Host-response-based gene signatures for tuberculosis diagnosis: A systematic comparison of 16 signatures. PLoS Med, 2019, 16(4): e1002786. doi:10.1371/journal.pmed.1002786. |
| [11] |
Walter ND, Miller MA, Vasquez J, et al. Blood Transcriptional Biomarkers for Active Tuberculosis among Patients in the United States: a Case-Control Study with Systematic Cross-Classifier Evaluation. J Clin Microbiol, 2016, 54(2): 274-282. doi:10.1128/JCM.01990-15.
pmid: 26582831 |
| [12] | Joosten SA, Fletcher HA, Ottenhoff TH. A helicopter perspective on TB biomarkers: pathway and process based analysis of gene expression data provides new insight into TB pathogenesis. PLoS One, 2013, 8(9): e73230. doi:10.1371/journal.pone.0073230. |
| [13] | Suliman S, Thompson EG, Sutherland J, et al. Four-Gene Pan-African Blood Signature Predicts Progression to Tuberculosis. Am J Respir Crit Care Med, 2018, 197(9): 1198-1208. doi:10.1164/rccm.201711-2340OC. |
| [14] | Rao M, Ippolito G, Mfinanga S, et al. Latent TB Infection (LTBI)-Mycobacterium tuberculosis pathogenesis and the dynamics of the granuloma battleground. Int J Infect Dis, 2019, 80S: S58-S61. doi:10.1016/j.ijid.2019.02.035. |
| [15] |
Mohammad-Rafiei F, Moadab F, Mahmoudi A, et al. Efferocytosis: a double-edged sword in microbial immunity. Arch Microbiol, 2023, 205(12): 370. doi:10.1007/s00203-023-03704-8.
pmid: 37925389 |
| [16] |
Martin CJ, Booty MG, Rosebrock TR, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe, 2012, 12(3): 289-300. doi:10.1016/j.chom.2012.06.010.
pmid: 22980326 |
| [17] |
Dallenga T, Repnik U, Corleis B, et al. M.tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe, 2017, 22(4): 519-530.e3. doi:10.1016/j.chom.2017.09.003.
pmid: 29024644 |
| [18] | Lee D, Cho M, Kim E, et al. PD-L1: From cancer immunotherapy to therapeutic implications in multiple disorders. Mol Ther, 2024, 32(12): 4235-4255. doi:10.1016/j.ymthe.2024.09.026. |
| [19] | 杨舒琪, 李锋. 程序性死亡受体1/程序性死亡-配体1抑制剂在结核病研究中的进展. 结核与肺部疾病杂志, 2025, 6(1): 94-101. doi:10.19983/j.issn.2096-8493.2024141. |
| [20] | Hu JF, Zhang W, Zuo W, et al. Inhibition of the PD-1/PD-L 1 signaling pathway enhances innate immune response of alveolar macrophages to mycobacterium tuberculosis in mice. Pulm Pharmacol Ther, 2020, 60: 101842. doi:10.1016/j.pupt.2019.101842. |
| [21] |
Jurado JO, Alvarez IB, Pasquinelli V, et al. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol, 2008, 181(1): 116-125. doi:10.4049/jimmunol.181.1.116.
pmid: 18566376 |
| [22] | Zhang W, Zhang Y, Liu Z, et al. PROS1-MERTK Axis Drives Tumor Microenvironment Crosstalk and Progression in Papillary Thyroid Microcarcinoma. Adv Sci (Weinh), 2025, 12(30): e13474. doi:10.1002/advs.202413474. |
| [23] |
Lumbroso D, Soboh S, Maimon A, et al. Macrophage-Derived Protein S Facilitates Apoptotic Polymorphonuclear Cell Clearance by Resolution Phase Macrophages and Supports Their Reprogramming. Front Immunol, 2018, 9: 358. doi:10.3389/fimmu.2018.00358.
pmid: 29545796 |
| [24] |
Nakayama K, Gazdoiu S, Abraham R, et al. Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem J, 2007, 401(1): 217-226. doi:10.1042/BJ20061135.
pmid: 16958618 |
| [25] |
Nakayama K, Qi J, Ronai Z. The ubiquitin ligase Siah2 and the hypoxia response. Mol Cancer Res, 2009, 7(4): 443-451. doi:10.1158/1541-7786.MCR-08-0458.
pmid: 19372575 |
| [26] | Qi J, Nakayama K, Gaitonde S, et al. The ubiquitin ligase Siah 2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways. Proc Natl Acad Sci U S A, 2008, 105(43): 16713-16718. doi:10.1073/pnas.0804063105. |
| [27] | Mehrotra P, Jamwal SV, Saquib N, et al. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog, 2014, 10(7): e1004265. doi:10.1371/journal.ppat.1004265. |
| [1] | Pharmaceutical Professional Branch of Chinese Antituberculosis Association. Expert consensus on therapy and drug monitoring clinical application of anti-tuberculosis drug (updated in 2025) [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 167-187. |
| [2] | Fu Xuwen, He Weiyaozhen, Li Xiang. Imaging manifestations of male reproductive system tuberculosis [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 188-196. |
| [3] | Su Wei, Bai Xinyu, Maiwulajiang Yimamu, Keyoumu Wubulikasimu, Diermulati Tusun, Lai Fuli, Mirenisha Abudurexiti, Xirizhati Mamuti, Zhou Linjun, Huang Fei. Survival status and influencing factors analysis of patients with rifampicin-resistant pulmonary tuberculosis in Kashgar Prefecture, Xinjiang Uygur Autonomous Region, 2013—2022 [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 197-205. |
| [4] | Wu Xiaoying, He Gang, Cai Xiaoting, He Liqian, Deng Hong, He Liyan. Analysis on the prognostic outcome model of elderly pulmonary tuberculosis patients with comorbidities based on the Chinese multimorbidity-weighted index [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 206-216. |
| [5] | Zhu Shiyu, Song Canlei, Zhang Rui, Hao Yanxing, Zhu Weitao, Gao Xia. Research on the construction of social support indicator system for tuberculosis based on Delphi method [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 217-222. |
| [6] | Xu Liangrun, Yang Mingying, Guo Yingwu, Wang Yun, Xu Jingjing, Hou Juyan, Ma Yunhong. A qualitative study on motivational factors and barriers to pulmonary tuberculosis treatment among initial treated smear-positive pulmonary tuberculosis patients and their families based on the Health Belief Model [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 223-229. |
| [7] | Liu Shuya, Zhang Ting, Wang Qingya, Fan Jun, Wang Ni. Analysis of close contacts screening situation and time changing trends of pulmonary tuberculosis patients in Chongqing from 2015 to 2024 [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 230-237. |
| [8] | Cai Liping, Chen Danping, Tang Jing, Jiang Sisi, Wu Ying, Wang Xiaomei. Construction of a discharge preparation service protocol for new drug-susceptible pulmonary tuberculosis patients based on the Timing Theory [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 238-246. |
| [9] | Lin Weibing, Shi Yiheng, Guo Yanling, Ma Shang, Yang Bin, Long Sibo, Zheng Maike, Sun Yong, Zhao Yan, Wang Guirong. Analysis of drug resistance patterns in 241 patients with lymph node tuberculosis [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 247-255. |
| [10] | Ji Yuanfei, Yu Xia, Wang Jue, Sun Mengyan, He Wei, Lyu Yan, Hou Dailun, Li Chenghai. The CT imaging features of Mycobacterium avium-intracellulare pneumonia and the diagnostic evaluation of in non-tuberculous mycobacterium pneumonia [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 256-263. |
| [11] | Su Ben, Qiu Lei, Zhou Rui, Chen Tao, Chen Yuxian, Cui Jie, Xia Liuyuan, Zhang Shaoyan, Lu Zhenhui. Analysis of usage characteristics of traditional Chinese medicine compound prescriptions for treating pulmonary tuberculosis based on the Chinese Patent Publication and Announcement Network [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 274-281. |
| [12] | Yu Haiwei, Li Qian, Feng Fengquan, Li Sha. Association analysis between anti-interferon-γ auto-antibodies and interferon-gamma release assay results [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 282-288. |
| [13] | Zhou Cang, Li Guoli, Jiang Hui, Shao Yan, Song Honghuan, Xue Hao, Ning Jingxian, Pan Yuchen, Fei Xinru, Sun Zheng, Chen Cheng, Zhu Limei, Liu Qiao. Evaluation of the diagnostic efficacy of AIMTB-fluorescent immunochromatography for screening latent tuberculosis infection [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 289-298. |
| [14] | Wang Xueyu, Chu Naihui, Nie Wenjuan. Research progress on the anti-Mycobacterium tuberculosis activity of sitafloxacin [J]. Chinese Journal of Antituberculosis, 2026, 48(2): 299-303. |
| [15] | Institute of Basic Research in Clinical Medicine, China Academy of Chinese Medical Sciences , Lishui Hospital of Traditional Chinese Medicine Affiliated to Zhejiang Chinese Medical University , Chinese Antituberculosis Association , Editorial Board of the Chinese Journal of Antituberculosis . Expert consensus on TCM syndrome differentiation, treatment principles, formulas, and herbs for latent tuberculosis infection [J]. Chinese Journal of Antituberculosis, 2026, 48(1): 1-8. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
 京公网安备11010202007215号
Total visitors: Visitors of today: Now online:
京公网安备11010202007215号
Total visitors: Visitors of today: Now online:
 This work is licensed under Creative Commons Attribution 3.0 License.
This work is licensed under Creative Commons Attribution 3.0 License.